
























理化学研究所 報道発表資料
http://www.riken.jp/pr/press/2014/20140310_1/
より、引用。
2014年3月10日
理化学研究所
新しいバイオインフォマティクス・ツール「ZENBU」を開発
―ゲノム上の数千もの転写活性を視覚化、解析し、データを共有―
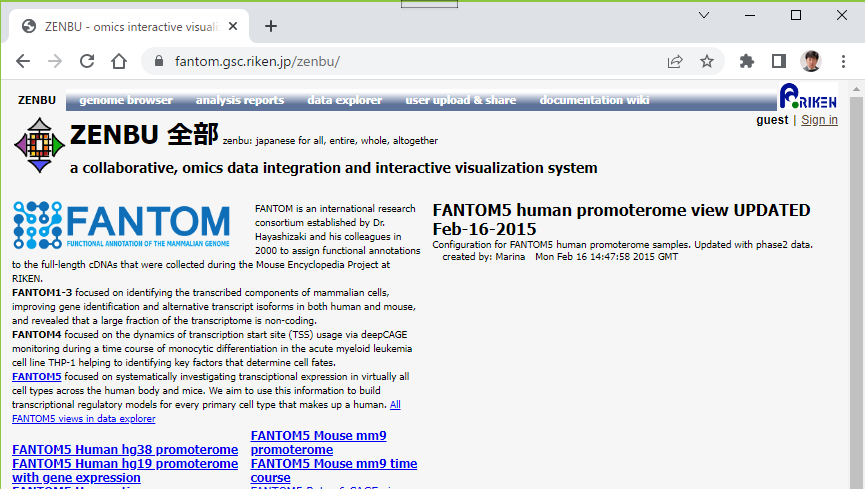
ZENBU 全部
http://fantom.gsc.riken.jp/zenbu/
●ポイント
○複雑なゲノム機能解析システムをWeb上で実現
○バイオインフォマティクスの専門家以外でも直感的に操作可能
○小規模の研究室でも大規模データセットの比較研究が容易に
●要旨
理化学研究所(理研、野依良治理事長)は、ゲノム配列のデータ解析とゲノムブラウザ[1]が連動したバイオインフォマティクス・ツール「ZENBU(ゼンブ)」を開発しました。ZENBUは誰でも無償で利用でき、次世代シーケンサー[2]から量産される大量の遺伝子発現情報の解析や視覚化、さらにはデータ間の比較を容易に行うことが可能です。これは、理研ライフサイエンス技術基盤研究センター(渡辺恭良センター長)機能性ゲノム解析部門(ピエロ・カルニンチ部門長)のアリスター・フォレストチームリーダー、ジェシカ・セヴェリン上級技師と、理研予防医療・診断技術開発プログラム(林崎良英プログラムディレクター)の川路英哉コーディネーターらとの共同研究グループによる成果です。
近年、ゲノム配列を高速解読する次世代シーケンサーの登場に加え、世界中の研究者が遺伝子機能を解析するさまざまな実験手法を利用するようにより、膨大な遺伝子データが生み出されています。しかし、得られた膨大なデータから生物学的意義を探り出す作業に多くの時間と労力がかかっています。バイオインフォマティクスは生物情報を扱う学術分野ですが、計算科学、生命科学両方の専門知識と経験を必要とすることから、研究者ですらその習得には多くの時間と労力を必要としていました。
共同研究グループは、この問題を解消するため、既存のゲノム情報視覚化ツールや解析ツールには実装されていなかった「膨大なデータをインタラクティブに自由自在に組み合わせて視覚化し、各種実験データにおけるさまざまな種類のデータ解析を実施できる」新たなゲノム機能解析ツールZENBUを開発しました。
ZENBUは無料で公開されており、インターネットを経由して利用する他、利用者それぞれのコンピュータにインストールして利用することも可能です。世界中の研究者が、公開後の実験結果をZENBUに登録し共有することで、最先端の研究がより素早く、共同で行われることを促進します。また、それぞれの研究者が自分の持つ実験データを最先端の研究データと比較することにより、新規の実験テーマの創出につながると期待できます。
本研究成果は、英国の科学雑誌『Nature Biotechnology』オンライン版(3月10日付け)に掲載されます。
●背景
近年、次世代シーケンサーの登場と、RNA-seq[3]やChIP-seq[4]、 DHS-seq[5]、CAGE[6]など、世界中の研究者が研究目的に応じた大量のゲノム配列データを一度に取得する革新的な遺伝子解析手法を利用するようになり、ゲノムの機能解析に関わるデータ生産量が急速に増加しています。ゲノムワイドな転写産物や転写因子結合領域の探索が可能になったことで、細胞機能や発生、がんなどの詳細な分子メカニズムの解明が期待されています。ただ、さまざまな手法から生み出される結果を統合し、比較することは、その分子メカニズムを理解するために必須ですが、日々生み出されてくる膨大な全てのデータセットを比較解析することは容易ではありません。既存のゲノム情報視覚化ツールやゲノムデータ管理ツールはいずれも、膨大なデータを視覚化するだけのものであったり、データ解析がリアルタイムで行われないものであったりするなど、1つのツールで全ての機能が実現可能なものはありませんでした。
●研究手法と成果
共同研究グループは、バイオインフォマティクスの経験が少ない利用者でも、次世代シーケンサーなどから産出される膨大なデータを可視化し解析に用いることができるツール「ZENBU(ゼンブ)」を開発しました。
既存のツールとZENBUとの大きな違いは、数千からなる膨大なデータをZENBU上で自由自在に組み合わせて、ゲノム上での特定の領域の発現シグナルをインタラクティブに視覚化、さらにリアルタイムで解析できることにあります。これらの機能が1つのツールで実現できるのが、ZENBUの革新的なポイントです。
現在、ZENBUには、ENCODE[7]及びFANTOM[8]コンソーシアムから6,000を超えるデータが登録されています。それぞれの研究者が自分の持つ実験データをそれらと比較研究することにより、新規の実験テーマの創出につながると期待できます。ZENBUは、次世代シーケンサーなどで得られた数百万以上のゲノム配列データを一度に登録できるように設計されており、ツールの名前であるZENBUの名はこれに由来しています。
●今後の期待
ZENBUは無料で公開されており、インターネットを経由した利用の他に、ソースコードを利用者それぞれのコンピュータにインストールし利用することも可能です。その場合、全てのZENBU利用者は相互接続することが可能であり、世界中の研究者が公開後の実験結果をZENBUに登録し共有することで、最先端の研究を共同で行う環境が構築されると期待できます。
●原論文情報
Jessica Severin, Marina Lizio, Jayson Harshbarger, Hideya Kawaji, Carsten O Daub, Yoshihide Hayashizaki, the FANTOM consortium, Nicolas Bertin, and Alistair RR Forrest. “Interactive visualization and analysis of large-scale NGS data-sets using ZENBU”. Nature Biotechnology, 2014 DOI:10.1038/nbt.2840
●発表者
○理化学研究所
ライフサイエンス技術基盤研究センター 機能性ゲノム解析部門 LSA要素技術研究グループ ゲノム情報解析チーム
チームリーダー アリスター・フォレスト (Alistair Forrest)
○独立行政法人理化学研究所
社会知創成事業 予防医療・診断技術開発プログラム
コーディネーター 川路 英哉 (かわじ ひでや)
●お問い合わせ先
独立行政法人理化学研究所
ライフサイエンス技術基盤研究センター
チーフ・サイエンスコミュニケーター 山岸 敦 (やまぎし あつし)
Tel: 078-304-7138 / Fax: 078-304-7112
●報道担当
独立行政法人理化学研究所 広報室 報道担当
Tel: 048-467-9272 / Fax: 048-462-4715
●産業利用に関するお問い合わせ
理化学研究所 社会知創成事業 連携推進部
●補足説明
[1] ゲノムブラウザ
ゲノム配列やゲノム機能に関する情報を分かりやすいように閲覧するシステム。
[2] 次世代シーケンサー
米国を中心に、従来のDNAシーケンサーが採用していたサンガー法とは異なる原理を採用することで、短いDNA配列をきわめて高速に解読する技術が開発され、活用できるようになった。シーケンサー開発分野は発展が目覚ましく、同じ次世代と言っても大きな性能差があるため、第2世代、第3世代と細分化されるようになっている。最近では、数千塩基以上の長いDNAの配列を決定できるシーケンサーも現れた。
[3] RNA-seq
RNA sequencingの略。組織や細胞で発現している全RNA(トランスクリプトーム)を解析する手法の1つ。mRNAやncRNAの断片的な配列情報(約50-125塩基)を網羅的に取得し、ゲノム配列と対応させることで、遺伝子発現量の定量や新たな転写配列の発見を行う。
[4] ChIP-seq
クロマチン免疫沈降法(chromatin immunoprecipitation:ChIP)と次世代シーケンサーを組み合わせることにより、転写因子などのDNA結合タンパク質や、特定の修飾を受けているヒストンが結合しているゲノム上の箇所を網羅的に解析する手法。
[5] DHS-seq
DNA分解酵素の一種DNaseIの高感受性部位(DNase-I-hypersensitive sites:DHS)であるゲノム領域を網羅的に解析する方法の1つ。細胞内のゲノムDNAはクロマチンと呼ばれる折り畳まれた構造をとるが、遺伝子の転写が活性化している領域などではその構造が崩れ、DNAが露出した状態になっていると考えられている。このような領域のDNAは、DNaseIを用いた酵素処理で50-100塩基対の断片として回収することができるため、次世代シーケンサーによる網羅的な配列決定が行える。ENCODEプロジェクトでは、ヒトゲノム上のDHSを全て記載することが提案されている。
[6] CAGE
Cap Analysis of Gene Expressionの略。理研が独自に開発した方法で、耐熱性逆転写酵素やcap捕捉法を組み合わせて転写物の5’末端の塩基配列を決定する実験手法。この塩基配列を読み取ってゲノム配列と照らし合わせて、どこから転写が始まっているかを調べることが可能。遺伝子の転写開始点をゲノムワイドに同定できる。
[7] ENCODE
ポストゲノム戦略として米国で立ち上げられたプロジェクトで、米国国立衛生研究所(National Institutes of Health:NIH)の国立ヒトゲノム解析研究所 (National Human Genome Research Institute)を中心として、2003年9月から正式に開始された計画。‘ENCODE’とは、Encyclopedia of Human DNA Elements(ヒトDNAの百科事典)から命名されており、完全解読されたヒトゲノム上に、遺伝子の機能を担う領域をすべて書き込んで、全ヒトゲノム (DNA)の百科事典を作成することを目指している。
http://www.genome.gov/10005107
[8] FANTOM
Functional ANnoTation Of Mammalian cDNAの略。哺乳動物(マウス)の遺伝子を網羅的に機能注釈することを主眼とする国際的研究コンソーシアム。2000年に、理研ゲノム科学総合研究センター 遺伝子構造機能研究グループ(旧・オミックス基盤研究領域、現・理研ライフサイエンス技術基盤研究センター 機能性ゲノム解析部門)が中心となって結成した。現在は活動範囲を拡大し、ヒト細胞の多様性を理解するため、さまざまな種類のヒト細胞を使って、転写開始部位の系統的マッピングに取り組んでいる。オーストラリア、シンガポール、スウェーデン、南アフリカ、イタリア、ドイツ、ギリシャ、スイス、英国、米国などを含む全世界の20カ国から、114の研究チームが参加している。
